
Drug Development and Regulatory Sciences

In a nutshell
Drug Development Science
Drug development science must go beyond basic efficacy to account for key factors such as:
- Drug disposition—This refers to how the body will interact with and handle a drug, accounting for factors such as absorption, distribution, metabolism, and excretion (ADME, also known as pharmacokinetics or drug disposition). No matter how well a drug works in the laboratory it will be ineffective if it cannot reach it’s target in the body or is metabolized and excreted too quickly. Conversely, it will carry a risk of toxic accumulation if it is not efficiently cleared.
- Drug-drug interactions—Many drugs are given in combination and nearly a third of American adults take five or more medications to manage chronic conditions. Recent U.S. Food and Drug Administration (FDA) guidance for safety testing of new dugs now includes determining their interactions with other drugs, including interference with metabolism or excretion.
- Genetic variations—Complex diseases such as cancer are varied in their underlying genetics. Thus drug development involves determining precisely which gene mutations are most effectively treated by which drugs, establishing therapeutic markers.
Department scientists create and apply innovative approaches to tackle these challenges and advance the science of drug development. These include:
- Discovering, detailing, and developing methods to predict the effect of transporters—portal-like proteins in cell and tissue membranes that take up or pump out small molecules—on drug disposition.
- Screening drug libraries for their effects on transporters, such as inhibition that can lead to drug-drug interactions.
- Partnering with the FDA to create and maintain the UCSF-FDA TransPortal database of pharmacologically relevant transporters to support the development of new drugs.
- Developing breast cell lines with specific cancer-related gene mutations. These cells are used to identify the most effective drugs for specific cancer genotypes.
- Demonstrating the feasibility of using computer models to simulate and predict the rate at which drugs will be cleared from the blood by the liver.
Regulatory Science
Research in regulatory science develops new tools, standards, and approaches to assess drug safety and efficacy.
Examples of department research and methods in drug development science include
Discovering the Role of Membrane Transporters in Drug Efficacy and Safety
Drug development must account for not just how a therapeutic molecule will effect its disease target, but how the body will handle the drug, that is, how it will absorb, distribute, metabolize, and excrete it (i.e., drug disposition). A drug that does not penetrate key membranes or is metabolized too quickly will not be able to affect its target, while drugs that are not efficiently cleared from the body can accumulate to toxic levels. The better that process of pharmacokinetics (PK) can be understood and predicted for a candidate molecule, the more efficient the drug development process.
Transporters are proteins in cell membranes that control the passage of certain molecules, including drugs, into and out of cells, tissues, and organs. There are uptake (absorptive) transporters that bind to and aid the entry of drug molecules and efflux transporters that pump them out. Thus transporters act as gatekeepers determining the extent and rate at which oral drugs enter the body via the intestine, are metabolized (e.g, in gut, liver), excreted (e.g, via the kidney), or reach target cells for therapeutic effect (e.g., brain, liver).
Department scientists pioneered and continue to play a leading role in understanding the key role of transporter proteins in determining drug disposition and safety. They help to develop predictive systems and data to aid in drug development, and they have contributed to regulatory guidance for drug developers related to testing drug candidates for transporter interactions.

Selected human transporter proteins for drugs and endogenous substances in membranes of (a) intestines; (b) liver; (c) kidney; and (d) the blood-brain barrier. Note based on arrow direction that some are uptake transporters, others are efflux. Source: Membrane transporters in drug development, Nature Reviews Drug Discovery, March, 2010, Vol. 9, p. 215-236, Giacomini KM et al.
Describing transporter-enzyme interplay and creating predictive systems to guide drug development
Researchers here were among the first to test the hypothesis that the poor oral bioavailability of some drugs (the extent that a pill-form drug reaches systemic circulation; a significant issue for both efficacy, as well as dose size and thus side effects risk) was not just the product of certain physicochemical properties (i.e, poor solubility in gastrointestinal fluids or low permeability through intestinal membranes), but was instead caused by transporter/enzyme interplay in the gut and liver.
Cellular, isolated organ, animal, and human studies here, initially focused on the immunosuppressant cyclosporine, which is vital to transplant patients. Researchers found that the drug readily passes through (permeates) the cells of intestinal lining (enterocytes), but is sent back into the gut by an efflux transporter, P-glycoprotein (P-gp). Each time the drug permeates the enterocytes it faces additional exposure to metabolism as a substrate of CYP3A enzymes, which act on more than one-half of all metabolized drugs. Thus, this complementary transporter/enzyme protective mechanism prevents 60 percent or more of the drug (which is notably a substrate for both proteins) from reaching the bloodstream where it acts.
Further studies, using drug-perfused rodent livers, found similar efflux transporter/enzyme interaction, but with the opposite effect, with drug not metabolized by CYP3A effluxed by P-gp, thus increasing drug concentrations in the blood.
These findings led department scientists to develop a way of predicting the relevance of transporters types and locations in drug disposition based on the U.S. Food and Drug Administration’s Biopharmaceutics Classification System (BCS). The latter categorizes drugs into four classes based on measures of their solubility and permeability [top left below]. Researchers here reviewed 130 BCS-classified compounds and noted that the major route of drug elimination for high permeability class 1 and 2 drugs in humans was metabolism, while low permeability class 3 and 4 drugs are eliminated unchanged via renal (urinary) and biliary excretion. They thus proposed a Biopharmaceutics Drug Disposition Classification System (BDDCS) [top right below], which predicts significant transporter effects and locations [see bottom row below].

- Upper left: The FDA’s Biopharmaceutics Classification System (BCS).
- Upper right: Biopharmaceutics Drug Disposition Classification System (BDCCS) where metabolism serves as permeability criteria.
- Bottom left: BDDCS notes transporter effects in the intestine following oral dosing.
- Bottom right: Generalized BDDCS transporter effects and locations. The system can guide testing of drugs for transporter interactions during early stages of drug development.
Sources: BDDCS applied to over 900 drugs, The AAPS Journal, Dec. 2011, vol. 13 (4), p. 519-547. Benet LZ, Broccatelli F, Oprea TI, and The Drug Transporter-Metabolism Alliance: Uncovering and Defining the Interplay, Molecular Pharmaceutics, Nov-Dec 2009, vol. 6 (6), p. 1631-1643, Benet LZ.
This disposition classification system can also help predict potential drug-drug interactions in the gut, liver, and brain—situations in which a transporter interaction with or inhibition by a drug may reduce a second drug’s enzymatic metabolism and/or clearance yielding toxic accumulation or reduced therapeutic effects—as well as the effect of high-fat meals on bioavailability due to their inhibition of gut transporters.
Researchers here have applied this drug disposition classification system to more than 900 already marketed drugs and developed a computational process for predicting BDDCS class from molecular structures. Such a system can be used in early drug discovery to optimize leads relative to efficacy and drug-drug interactions.
Providing predictions, testing, and regulatory guidance on transporters and drug-drug interactions
Each year more than 2 million people in the United States suffer severe adverse drug reactions that can result in death and disability. About one-fourth of those reactions are caused by the interaction of drugs taken at the same time.
It has long been known that some of these problems occur when one drug inhibits enzymes that would normally metabolize a second drug, causing the latter to accumulate to toxic levels. As a result, drugs are tested for such effects before approval.
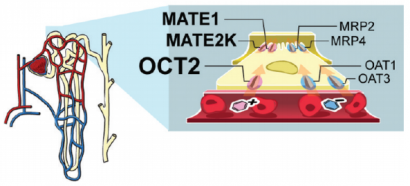
OCT2 and other drug transporter proteins (at right) in the cell membranes of kidney tubules (at left). Tubules interface with capillaries to remove drugs for excretion in urine.
Department research helps to focus attention on drug-drug interactions (DDIs) that occur due to drugs inhibiting transporters vital to excretion. For example, in the kidneys, where most prescription drugs are cleared from the body. Studies here have screened hundreds of drugs against kidney transporters, such as OCT2, which control the first step of kidney clearance for most drugs. This includes metformin, the most widely used medication for type 2 diabetes. One study found six drugs which, at clinically relevant concentrations, so significantly inhibited OCT2 function that they merited clinical studies for generating DDIs.
Indeed, department researchers as part of the International Transporter Consortium—a collaborative group of scientists from academia, industry, and the U.S. Food and Drug Administration (FDA)—informed FDA guidance to drug developers on evaluating new drugs for drug-drug interactions. The FDA recommends in vitro testing to quantify the contribution of transporters to drug disposition, establishing if a drug is a substrate or inhibitor of particular transporters. For example, if a new drug inhibits a transporter in vitro at therapeutically relevant concentrations, then that may cause drug-drug interactions and may require further in vitro or in vivo studies to determine its safety.
Molecular docking against transporters to aid drug safety and discovery
Drug molecule interactions with transporters can be positive, for example if their structures make them the substrates of influx transporters that deliver them to therapeutic targets, say, across the blood-brain barrier or negative if an efflux transporter prevents a drug from being absorbed in the intestine.
A key goal for drug development is to understand which small molecule drug structures interact with transporters in what ways (structure-activity relationships) so that such interactions can be predicted for more efficient drug development.
One strategy used by department scientists to map drug interactions with transporters is structure-based ligand discovery, including tapping molecular docking computer programs (e.g., UCSF DOCK) that virtually screen and rank large libraries of small molecules ligands for the relative strength with which they bind to a target protein—in this case, a drug-relevant transporter. High-ranking ligands are then experimentally tested to confirm transporter binding and determine their effects (i.e., inhibitor, substrate). Molecular structures can potentially be used to identify similar small molecule drugs that interact with the transporters in a therapeutic manner or screen candidates for negative interactions.
A major challenge for this computational docking approach is the difficulty in obtaining experimentally determined, high-quality structures of mammalian transporters due to factors such as their dynamic flexibility and the difficulties encountered in crystallizing the large and poorly soluble (hydrophobic) membrane proteins for x-ray diffraction analysis. In fact, membrane proteins such as transporters comprise only about 1 percent of the determined structures in the worldwide Protein Data Bank.
Thus research here taps the department’s notable strength in comparative modeling—which determines the 3-D structures of an unknown target proteins in part by aligning its carbon backbone with that of a determined template protein that is evolutionarily related as reflected in sequence similarity (homologous).
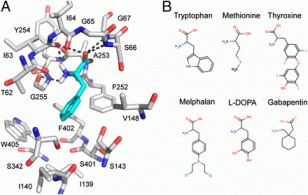
Stick model of the predicted structure of the human LAT-1 transporter protein and ligand-binding mode based on modeling from bacterial homologs. The ligand (cyan) is phenylalanine, a known LAT-1 substrate.
Department studies have, for example, used the X-ray crystallography determined structure of distantly related bacterial homologs to construct a comparative model of the LAT-1 human transporter. LAT-1 transporters are particularly numerous (highly expressed) at the blood brain barrier where they provide for absorption of nutrients (essential amino acids) as well as drugs such as L-dopa to treat Parkinson’s Disease and gabapentin to treat seizures, making them targets for central nervous system drug delivery. These transporters are also increased in many cancers in which they import nutrients and pro-proliferative signaling molecules to tumor cells, thus making them cancer treatment targets.
The LAT-2 model was used for virtual screening against more than 19,000 small molecules followed by experimental validation of select highly ranked ligands that had novel chemical structures and pharmacological effects. The study found four previously unknown LAT-1 ligands, including two drugs known for crossing the blood brain barrier to treat the central nervous system (CNS) and two that were found to inhibit LAT-1 enabled cancer cells. Intriguingly, one of the latter failed in cancer treatment trials because of toxic CNS-related side effects, providing an archetypal example of the value of such virtual screening against transporters in drug development.
Identifying genetic markers to match the most effective drugs to patients
Cancer is not a uniform disease but rather can result from mutations in a range of key genes, including oncogenes and tumor suppressor genes, which then promote or allow its abnormal and unchecked cellular proliferation. Therefore a key goal in cancer drug development is to discover genetic markers that predict and modulate response to small molecule therapeutics. This will allow for personalized medicine, matching the drugs to specific cancer genotypes.
Department scientists have developed a panel of 50 human epithelial cell lines derived from normal breast tissue, with either individual activated oncogenes or deletions/alterations to key tumor suppressor genes that are commonly seen in cancers. Researchers here are using high-throughput screening to rapidly measure cell proliferation in response to drugs They are identifying which oncogenes affect the cells’ response to individual drugs as well as different combinations of drugs. This approach not only detects the most effective drugs for specific cancer cell genotypes but also which genes are conferring drug resistance, thus guiding the development of more potent therapies.
Computer models provide insights into drug clearance by the liver
Drug development can be made faster and less expensive through computer models that simulate how the body handles drug molecules or even how drugs interact with various components on or within key cells. For example, experiments on a computer-simulated (in silico) liver or cultured hepatocytes, which comprise most of the liver and metabolize drugs, seek to predict changes in drug clearance from the blood when multiple drugs are co-administered for different health or disease scenarios.
Such in silico screening of molecules for clearance patterns in early drug development could save time and resources spent on unworkable drug candidates.
In some experiments department scientists use virtual analogs of mice and in others analogs of hepatocyte cultures. In both cases, in silico hepatocytes (ISHs) simulate metabolism of virtual drug molecules. The ISHs include modular components known to influence hepatocyte intrinsic clearance, including transporters (See top section above.) and different types of drug metabolizing enzymes, such as the cytochrome P450 superfamily.
Computational experiments put thousands of virtual drug molecules into in silico cultures made up of thousands of ISHs. After comparing simulation results with those of in vitro experiments, aspects of the model are adjusted to increase its credibility. Researchers here have demonstrated feasibility by comparing clearance predictions from virtual experiments for a variety of in silico compounds, having particular properties, to actual wet-lab measurements of their real world counterparts.
Department scientists envision this work, along with that of others, leading to more advanced, increasingly credible analogs engineered to function as virtual patients that have been tailored (simplified) to represent a particular individual’s health (or disease) issues. Experiments will be conducted on copies of that virtual patient to explore outcomes of several different multi-drug treatment protocols, for which evidence suggests that each may be beneficial.
Examples of department research and methods in regulatory science include
Department scientists co-lead the FDA-funded UCSF-Stanford Center for Excellence in Regulatory Science and Innovation (UCSF-Stanford CERSI), the first such federally funded regulatory science center in the Western United States. The center taps department expertise in quantitative pharmacology—integrating diverse data from different stages of drug development via informatics and computer models—to improve preclinical and clinical safety and efficacy studies.